


Ein neues Gen für Elias

Wenn Babys mit ihren Beinchen strampeln und dabei lächeln, kommt das frischgebackenen Eltern im Rausch der Glückshormone immer wie ein kleines Wunder vor. Dass der inzwischen einjährige Elias lächeln und strampeln kann, ist jedoch tatsächlich eines, denn der kleine Junge kam mit Spinaler Muskelatrophie (SMA) zur Welt. Ohne Behandlung sterben die meisten Kinder innerhalb kurzer Zeit an dem angeborenen und schnell fortschreitenden Muskelschwund. Dass Elias lebt und seiner Familie auch in Zukunft viel Freude machen wird, verdankt er der Erweiterung des Neugeborenenscreening um die Krankheit SMA, den Ärztinnen und Ärzten, Forschenden, Pflegenden und Mitarbeitenden im Labor des Universitätsklinikums Heidelberg (UKHD) sowie einer modernen Gentherapie.
Als Elias im Oktober 2021 in Bad Mergentheim zur Welt kommt, deutet nichts darauf hin, dass mit dem kleinen Jungen etwas nicht in Ordnung sein könnte. Alle Untersuchungen im Krankenhaus sind unauffällig und die Eltern werden mit einem gesunden Kind entlassen. Er bezaubert alle um sich herum mit seiner munteren Art und den großen braunen Augen. Zu Hause erwarten ihn die Geschwister begeistert.
Wie beim Neugeborenenscreening üblich, hat Elias Kinderarzt in den ersten Lebenstagen noch im Krankenhaus etwas Blut aus der Ferse entnommen und tropft es auf das Filterpapier einer sogenannten Trockenblutkarte. Per Post geht das Blutkärtchen ins Labor nach Heidelberg und wird dort auf Auffälligkeiten untersucht. Täglich treffen dort Kärtchen aus Rheinland-Pfalz, Baden-Württemberg, dem Saarland und Teilen Nordrhein-Westfalens ein. Im Juli 2021 wurde das Neugeborenen-Screening des Universitätsklinikums Heidelberg in einer Pilotphase um zwei Erbkrankheiten erweitert: Spinale Muskelatrophie (SMA) sowie Sichelzellerkrankung. Seit September 2021 sind beide Erkrankungen deutschlandweit ins Neugeborenen-Screening integriert worden. Ein Glücksfall für Familie Wehr, wie sich später zeigen wird.
„Ziel des Screenings ist es, die Diagnose so früh wie möglich zu stellen, um rechtzeitig mit der Behandlung zu beginnen. Denn vor allem bei SMA zählt jeder Tag, um irreversible Schäden abzuwenden“, sagt Professor Dr. Georg F. Hoffmann, Direktor des Zentrums für Kinder- und Jugendmedizin am UKHD. Er ist verantwortlich für dieses und weitere erfolgreiche Screening-Pilotprojekte am Standort Heidelberg. Diese werden seit über 20 Jahren von der Dietmar Hopp Stiftung mit bislang rund 16 Mio. Euro gefördert.
Elias ist mit seinen Eltern Jessica und Martin bereits seit einer Woche zu Hause als das Telefon klingelt. Das Krankenhaus Bad Mergentheim meldet sich bei der Familie mit einer schlimmen Nachricht. Der Bluttest aus dem Neugeborenen-Screening hat gezeigt, dass Elias an SMA leidet. Ein Schock für die Eltern. „Wir konnten anfangs nicht glauben, dass unser Sohn plötzlich einen tödlichen Gendefekt haben soll. Von einer Sekunde auf die andere wurde aus einem gesunden Kind ein todkrankes Kind“, erinnern sich die Eltern an den Tag der Diagnose.
Innerhalb von drei Stunden sollen sie zum Termin mit Dr. Andreas Ziegler in Heidelberg sein. Bei einer Fahrzeit von zwei Stunden von Weikersheim nach Heidelberg am Freitag eine sportliche Herausforderung. Und auch die Zwillinge mussten noch schnell zur Oma gebracht werden. Aber die Familie schafft alles und sitzt wenig später bei Dr. Ziegler zum Beratungsgespräch und bespricht das weitere Vorgehen. Dr. Ziegler ist Oberarzt der Sektion Neuropädiatrie und Stoffwechselmedizin des Zentrums für Kinder- und Jugendmedizin und forscht seit Jahren im Bereich der spinalen Muskelatrophie.


„Wir konnten anfangs nicht glauben, dass unser Sohn plötzlich einen tödlichen Gendefekt haben soll. Von einer Sekunde auf die andere wurde aus einem gesunden Kind ein todkrankes Kind.“
Elias hatte Glück, dass zu seiner Geburt das Neugeborenenscreenings nun auch bundesweit um die Krankheit SMA erweitert wurde.
Nun ist rasches Handeln gefragt. Denn unbehandelt verläuft SMA noch vor dem zweiten Lebensjahr tödlich. „So selten ist die Erkrankung gar nicht, jedes Jahr sind circa 100 Neugeborene in Deutschland davon betroffen. Eines von 7500 Neugeborenen hat SMA“, sagt Dr. Ziegler. Bei SMA-Patientinnen und -Patienten funktioniert wegen eines Gendefekts an einer bestimmten Stelle des Erbguts die Kommunikation zwischen Nervenzellen im Rückenmark und der Muskulatur nicht. Spinale Muskelatrophie wird nur vererbt, wenn beide Eltern Träger eines defekten SMN1-Gens sind. Die Neuronen im Vorderhorn des für die Motorik zuständigen Rückenmarks gehen nach und nach zugrunde. Die Bewegungssignale aus Gehirn und Rückenmark werden aufgrund der fehlenden Schaltstellen nicht mehr an die Muskulatur weitergegeben, und die Patienten verlieren immer mehr die Möglichkeit, sich zu bewegen. Bei den früh einsetzenden Formen der SMA schreitet der Muskelabbau rasant voran und breitet sich schließlich auch auf die Atemmuskulatur aus.

„Wir haben das große Glück, dass Elias noch keine Einschränkung durch die Erkrankung hatte. Und auch jetzt verlaufen alle Kontrolluntersuchungen unauffällig. Wir sind sehr dankbar und glücklich, dass die Erkrankung bei unserem Sohn so früh entdeckt und behandelt wurde.“

Für Elias, der jene früh einsetzende Form der SMA hat, kommt die Hilfe rechtzeitig. An seinem 16. Lebenstag erhält er eine Genersatztherapie in Form des Medikaments Zolgensma – als einmalige Infusion. „Dabei wird über ein modifiziertes Virus eine intakte Kopie des defekten SMN1-Gens über den Blutkreislauf in den Körper und damit auch die betroffenen Neuronen eingeschleust. Das sichert das Überleben der Nervenzellen“, erklärt Dr. Ziegler.
Man geht derzeit davon aus, dass das Ersatzgen lebenslang aktiv bleibt. „Da die Therapie aber noch sehr neu ist, gibt es noch keine Langzeitergebnisse“, sagt der Mediziner. Als Nebenwirkungen treten bisweilen vorübergehende Entzündungen verschiedener Organe auf, die man jedoch gut behandeln kann. In seltenen Fällen kann es zu Leberversagen kommen.
Elias bleibt davon verschont und übersteht die Behandlung und den einwöchigen Aufenthalt in der Kinderklinik am UKHD gut: „Er hat die Behandlung gut vertragen“, freut sich Dr. Ziegler.

Im Stoffwechselzentrum Heidelberg werden jährlich die Proben von mehr als 140.000 Neugeborenen aus Baden-Württemberg, Rheinland-Pfalz, Nordrhein-Westfalen und dem Saarland untersucht.
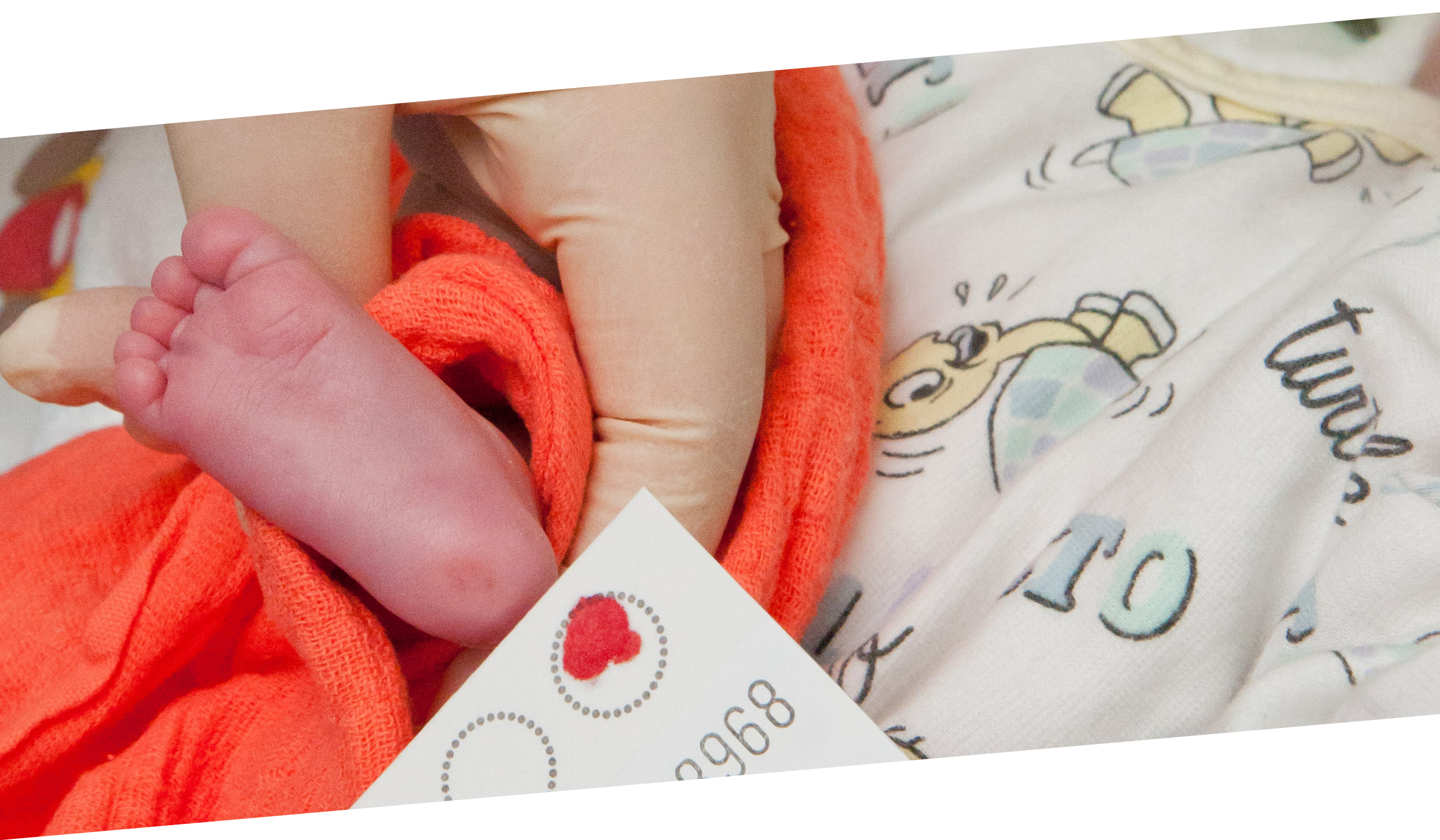
„Ohne Therapie hätte Elias inzwischen mit Einschränkungen zu kämpfen und seine Lebenserwartung wäre begrenzt. Da bei ihm der Gendefekt bereits sehr früh festgestellt und behandelt wurde, hat er Aussicht auf ein normales Leben“, sagt Dr. Andreas Ziegler.
Elias geht es prächtig. Dr. Ziegler und Elias Eltern sind zufrieden mit seiner Entwicklung. Alle vier Monate wird er in Heidelberg untersucht, um den Langzeitverlauf im Rahmen der Therapie besser verstehen zu können, denn er ist das erste Kind mit SMA, das zum Zeitpunkt der Gentherapie noch keine Symptome der SMA zeigte.
„Jeden kleinen Fortschritt und jede positive Entwicklung feiern wir“, sagen die Eltern. „Wir sind sehr dankbar und glücklich, dass die Erkrankung so früh entdeckt und behandelt wurde.“
Am Universitätsklinikum Heidelberg wird auch in Zukunft intensiv geforscht im Bereich der Sekundärprävention von Erkrankungen im Kindesalter …
… durch Aufnahme weiterer Piloterkrankungen in das Neugeborenen-Screening und deren frühe und effiziente Behandlung mit neuartigen Arzneimitteln.
Mit finanzieller Unterstützung durch die Dietmar Hopp Stiftung wird die Aufnahme weiterer Zielkrankheiten in das Neugeborenenscreening evaluiert. Die Stiftung unterstützt darüber hinaus den Aufbau einer professionellen Infrastruktur für die klinische Forschung im Kindes- und Jugendalter am UKHD in Form des sog. pädiatrisch klinisch-pharmakologischen Studienzentrums (paedKliPS), das gemeinsam mit der Abteilung für klinische Pharmakologie und Pharmakoepidemiologie am Campus aufgebaut wird.
In einem weiteren zukunftsweisenden Versorgungsforschungsprojekt werden das Zentrum für Kinder- und Jugendmedizin und die medizinische Klinik V, der Abteilung für Onkologie und Hämatologie, gemeinsam von Heidelberg aus innovative Strukturen für die Anwendung der sogenannten Arzneimittel für neuartige Therapien, der ATMPs, in Deutschland, schaffen, ein Projekt, das vom Innovationsfond der gesetzlichen Krankenkassen in den nächsten Jahren mit insgesamt 13,6 Millionen Euro gefördert wird.