


Frau Schmidts Traum vom Schnorcheln
Wer eine seltene Erkrankung hat, trägt oft eine besonders schwere Last. Das Zentrum für Seltene Erkrankungen am Universitätsklinikum Heidelberg verbindet 18 Fachzentren und bietet Betroffenen Hoffnung und Hilfe. Maren Schmidt* ist eine von ihnen. Die 26-Jährige hat das Loeys-Dietz-Syndrom, ein seltener Gendefekt mit einem hohen Risiko für lebensbedrohliche Aneurysmen der Hauptschlagader. Schon mehrfach hat das Team des Zentrums ihr das Leben gerettet. *Name geändert
Als Maren Schmidts Nieren ausfallen, rauschen jede Nacht Flüsse durch ihre Träume. „Ich konnte nur noch an Wasser denken“, erzählt die 26-Jährige. In ihren Träumen hält sie die Füße ins kühle Nass oder schwimmt im Meer. In der Realität muss sie zur Dialyse und darf nicht viel trinken. Durst ist ihr ständiger Begleiter. Zur Ablenkung schaut sie sich Dokumentationen rund ums Tauchen an. So entsteht ihr größter Wunsch: einmal mit Meeresschildkröten zu schwimmen. Doch derzeit darf sie nicht verreisen, verbringt viele Monate im Krankenhaus. Denn Maren Schmidt hat das äußerst seltene Loeys-Dietz-Syndrom.

Unklare Diagnose im Kindesalter
Ihre Krankheitsgeschichte beginnt im Alter von 13 Jahren. Damals wird sie wegen starker Bauchschmerzen ins Universitätsklinikum Heidelberg eingeliefert. Man vermutet zunächst eine gewöhnliche Blinddarmentzündung. Obwohl bei Maren Schmidt schon im frühen Kindesalter die angeborenen seltene Gefäßerkrankung Klippel-Trénaunay-Weber-Syndrom vermutet wurde, war sie bis auf einige Krampfadern und eine Umfangsvermehrung im rechten Bein bis dato kerngesund. „Bei den Untersuchungen kam heraus, dass ich keinen entzündeten Blinddarm, sondern ein sogenanntes Aortenaneurysma hatte“, erinnert sie sich. Damit ist eine Ausdehnung der Hauptschlagader im Bauchraum gemeint. Ein Aortenaneurysma ist in diesem Alter sehr ungewöhnlich.
Damals ist noch keine operative Behandlung nötig, doch als Maren Schmidt 15 ist, muss sie in der Gefäßchirurgie operiert werden. Die Aorta droht bei einem Durchmesser von mehr als fünf Zentimetern – normal in diesem Lebensalter sind 1,5 Zentimeter – zu reißen. Das wäre lebensgefährlich.
„Wir haben das erkrankte Gefäßsegment durch eine Kunststoff-Prothese ersetzt“, erzählt ihr behandelnder Arzt Professor Dr. Dittmar Böckler, Ärztlicher Direktor der Klinik für Gefäßchirurgie und Endovaskuläre Chirurgie am Universitätsklinikum Heidelberg (UKHD). „Danach hatte ich erst einmal zehn Jahre Ruhe“, ergänzt seine Patientin.

Professor Dr. Philipp Erhart

Zweimal verschlechtert sich Maren Schmidts Zustand so dramatisch, dass sie mit dem Rettungshubschrauber abgeholt und nach Heidelberg geflogen wird.
Operationen in Serie
Nach der Operation kommt sie alle ein bis zwei Jahre zur Kontrolle nach Heidelberg. Bei einem dieser Besuch in Heidelberg zeigt sich, dass auch ein weiterer Teil der Aorta ersetzt werden muss. Im Herbst 2023 wird die junge Frau erfolgreich operiert. Doch kaum ist sie zu Hause, jagt ein Unglück das nächste: Ihre Nieren und die Leber versagen, und ihr Herz schlägt nur noch sehr schwach. „Ich wurde mit dem Hubschrauber abgeholt und nach Heidelberg geflogen“, erinnert sie sich. In der Kardiologie (Leitung: Prof. Dr. Norbert Frey) wird sie erfolgreich behandelt und später nach Hause entlassen. Sie muss allerdings ab jetzt zur Dialyse.
Eine Woche später erleidet sie eine sogenannte Aortendissektion – dabei reißt die innerste Wandschicht der Hauptschlagader. Maren Schmidts Zustand ist lebensbedrohlich, wieder fliegt der Helikopter sie nach Heidelberg, wo sie diesmal vom herzchirurgischen Team (Leitung: Prof. Dr. Matthias Karck) notoperiert wird. Seitdem verlässt sie das Krankenhaus für mehrere Monate nicht. Alle paar Tage muss sie für geplante Folgeoperationen in den OP, mit der Wundheilung gibt es Probleme. Ihre Nieren arbeiten zum Glück wieder besser; die Dialysebehandlungen gehören der Vergangenheit an.
Stark vernetzt
Um eine umfassende und effektive Therapie sicherzustellen, arbeiten Expertinnen und Experten verschiedener medizinischer Disziplinen eng zusammen. Nur so kann es gelingen, die komplexen Symptome des Loeys-Dietz-Syndroms in Schach zu halten. Denn eine Heilung gibt es bislang nicht.
Interdisziplinäre Zusammenarbeit
Erst seit einigen Wochen weiß Maren Schmidt, dass sie nicht am Klippel-Trénaunay-Weber-Syndrom leidet, sondern das Loeys-Dietz-Syndrom hat – dank einer genetischen Untersuchung, die das Fachzentrum für Seltene Gefäßerkrankungen unter Leitung von Professor Dr. Philipp Erhart in Zusammenarbeit mit dem Institut für Humangenetik (Leitung: Professor Dr. Christian Schaaf) durchführt. Das Loeys-Dietz-Syndrom ist eine sehr seltene genetische Erkrankung: „Eine von einer Million Personen ist betroffen“, sagt Professor Erhart. Am Zentrum für Seltene Gefäßerkrankungen hat er dieses Syndrom erst bei einer weiteren Patientin diagnostiziert. Es betrifft das Bindegewebe und kann Probleme mit den Blutgefäßen, dem Herz-Kreislauf-System und dem Skelett verursachen.
Lebensbedrohliche Komplikationen sind Gefäßeinrisse, denn durch den Gendefekt sind die Wände der Arterien geschwächt. „Bei Frau Schmidt sind auch andere Organsysteme erkrankt, so dass Kardiologen, Nephrologen, Gynäkologen, Radiologen und Herzchirurgen an der Behandlung beteiligt waren“, erklärt Professor Erhart. In regelmäßigen interdisziplinären Fallkonferenzen besprechen die Fachleute das weitere Vorgehen. Da die zu behandelnden Arterien sehr vulnerabel sind, müssen die operativen Eingriffe wohlüberlegt sein und intensiv vorbereitet werden, sagt Erhart. So wird sichergestellt, dass die Eingriffe effizient und komplikationslos verlaufen.
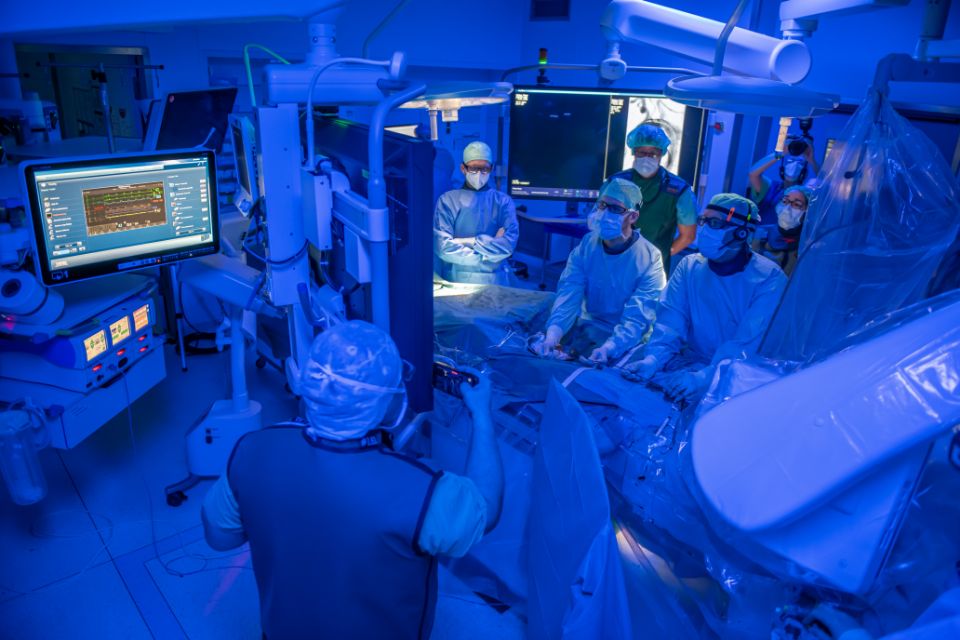
Da beim Loeys-Dietz-Syndrom mehrere Organe erkrankt sind, bedarf es nicht nur während der Operation, sondern auch davor und danach einer interdisziplinären Zusammenarbeit.
Im Video stellt sich die Klinik für Gefäßchirurgie und Endovaskuläre Chirurgie zusammen mit Pflege und ihren Partnern aus Chirurgie und Anästhesiologie am UKHD vor.
Aktuell stößt die Medizin bei der Behandlung des Loeys-Dietz-Syndroms an ihre Grenzen, bedauern die Mediziner: „Eine kausale Therapie dieses Gendefekts existiert leider nicht. Man kann die Erkrankung nur chirurgisch behandeln und durch lebenslange Nachsorgen rechtzeitig nach neuen Gefäßveränderungen Ausschau halten.“ Im Bereich der Brustschlagader erhält Maren Schmidt in Kürze noch eine sogenannte Endoprothese, um das Fortschreiten der Erweiterung im letztverbliebenen unbehandelten Abschnitt der Brustschlagader zu stoppen und einen Riss bzw. Ruptur an dieser Stelle zu verhindern. „Das ist ein minimalinvasiver Eingriff, ähnlich einer Herzkatheteruntersuchung, erklären die Gefäßchirurgen Böckler und Erhart.
Sie sei eigentlich ein positiver Mensch, sagt Maren Schmidt. Doch die vielen Monate im Krankenhaus gehen auch an ihr nicht spurlos vorbei: „Das macht etwas mit einem.“ Durch die vielen Operationen ist sie oft müde. Um endlich mal wieder ein Buch zu lesen oder einen ganzen Film zu schauen, dafür reicht ihre Konzentration noch nicht aus. Die langen Krankenhaustage vertreibt sie sich mit Origami, Instagram und Dokumentationen. Kraft geben ihr die Eltern und die beiden Brüder, die jeden Tag zu Besuch kommen: „Wenn ich meine Familie nicht hätte, wäre ich nicht so optimistisch.“
Das Zentrum für Seltene Erkrankungen vereinigt die Zentren für …
- … Amyloidose
- … Seltene Blutkrankheiten
- … Echinokokkose und seltene Tropenerkrankungen
- … angeborene Endokrinopathien
- … seltene Herzerkrankungen
- … seltene kranio-orofaziale Erkrankungen
- … seltene Lungenerkrankungen
- … Mukoviszidose
- … seltene Nierenerkrankungen
- … seltene orthopädische Erkrankungen
- … seltene entzündlich-rheumatische Erkrankungen
- … chronischen Singultus
- … angeborene Stoffwechselerkrankungen
- … syndromale Entwicklungsstörungen
- … seltene Tumorerkrankungen
- … seltene neurologische Erkrankungen
- … seltene Gefäßerkrankungen
- … seltene angeborene Fehlbildungen des Verdauungstraktes
Mit Schildkröten schnorcheln
Das Ergebnis des Bluttests habe sie überrascht und auch ein wenig erschreckt, sagt Maren Schmidt. „Ich wusste ja nicht, welche neuen Einschränkungen jetzt kommen.“ Weiterhin muss die junge Frau darauf achten, dass sie nicht zu viel Druck auf ihre Gefäße ausübt. Ausdauersportarten wie Schwimmen, Wandern und Fahrradfahren sind beim Loeys-Dietz-Syndrom in Absprache mit den behandelnden Kardiologen empfehlenswert, aber Kontaktsportarten wie z.B. Fuß- oder Handball soll sie meiden; auch tief tauchen darf sie wegen des Unterwasserdrucks nicht.
Eines freut den Wasserfan sehr: „Schnorcheln ist erlaubt.“ Wenn sie das Krankenhaus wieder verlassen darf und auch die Reha hinter sich hat, möchte sich Maren Schmidt ihren großen Traum erfüllen und mit Schildkröten im Mittelmeer schwimmen. Am liebsten in Griechenland, der alten Heimat ihrer Mutter. Dass dieser Traum wahr wird, daran arbeiten sie und das Team des Zentrums am Universitätsklinikum Heidelberg jeden Tag.


„Eine Diagnose ist auch für die Psyche wichtig“